



Vanessa Denny, MD1; Brian O’Connor, MD2; Saira Siddiqui, MD1
Affiliations:
1. Goryeb Children’s Hospital, Atlantic Health System, Morristown, NJ
2. Robert Wood Johnson University Hospital
Case Published in the Journal of Cardiovascular Magnetic Resonance: Click here for the link
Click here for PubMed Reference to Cite this Case
Clinical History
A 16-year-old highly athletic boy with no significant past medical history or family history, presented with recurrent presyncope. During a basketball game, he developed an hour-long episode of dizziness, pallor, and diaphoresis. EMS obtained a 12-lead electrocardiogram and found him to be in monomorphic ventricular tachycardia at a rate of 250 bpm, and successfully cardioverted him. He was admitted to an outside hospital where a limited echocardiogram reportedly demonstrated normal cardiac anatomy and function (Figure 1). An EKG obtained during a brief episode of non-sustained ventricular tachycardia demonstrated origin from the right ventricular outflow tract (Figure 2 top). His baseline EKG was normal with a RSR’ pattern in V1 and V2 (Figure 2 bottom). He was referred for cardiac MRI (CMR) for further evaluation.
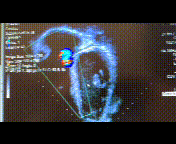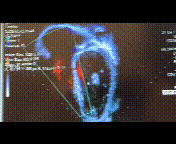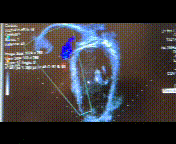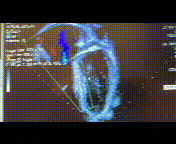
Figure 1: Apical 4 chamber view on echocardiogram.
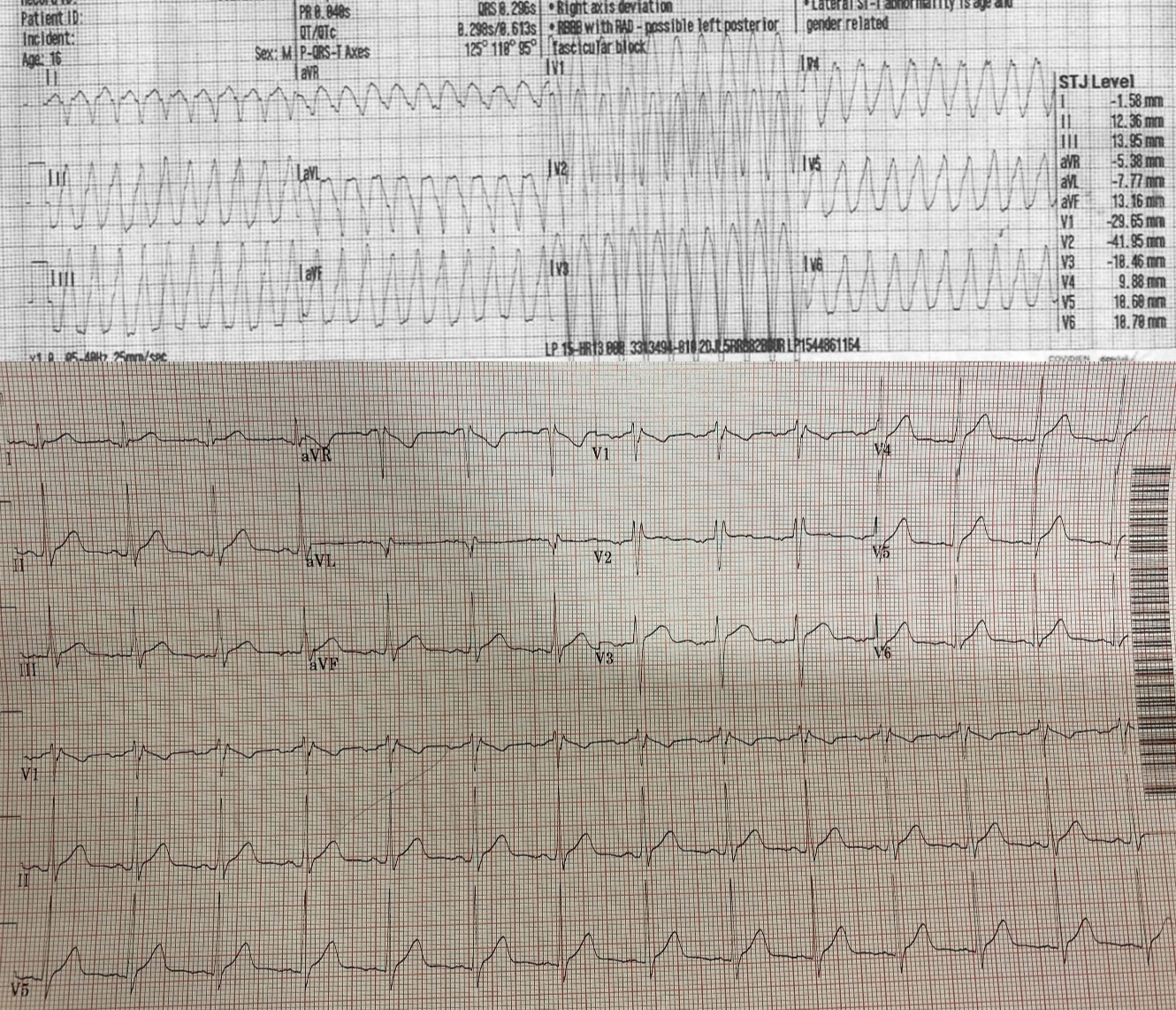
Figure 2: 12 lead EKG demonstrating non-sustained ventricular tachycardia originating from the right ventricular outflow tract (top). Baseline EKG (bottom).
CMR Findings
CMR with SSFP cine imaging, black blood T1-weighted imaging with and without fat saturation, T1 parametric mapping, and late gadolinium enhancement imaging was performed. 4-chamber SSFP cine images demonstrated right ventricular dilation with right ventricular dyskinesia (Figure 3). The left ventricular lateral wall appeared irregular (Figure 3). 2-chamber SSFP right ventricular cine demonstrated right ventricular microaneurysms along the lateral free wall, consistent with the “accordion sign” (Figure 4, right). 2-chamber SSFP left ventricular cine demonstrated normal left ventricular systolic function (Figure 4, left). The short axis cine demonstrated a dilated right ventricle (indexed right ventricular end-diastolic volume of 155 ml/m2) with globally mildly reduced systolic function, with an ejection fraction of 42%. There was normal left ventricular systolic function with an ejection fraction of 55% with no significant wall motion abnormalities (Figure 5). The left ventricle was mildly dilated with an indexed left ventricular end-diastolic volume of 122 ml/m2 (Figure 5). T1-weighted fast-spin echo (FSE) black blood imaging with and without fat saturation suggested possible epicardial fat infiltration of the lateral left ventricle (Figure 6). Parametric mapping with T1 mapping demonstrating normal mean native T1 signal. On late gadolinium enhancement imaging, there was subepicardial to mid-wall enhancement at the left ventricular apical lateral segment (Figure 7).

Figure 3: 4-chamber SSFP cine.
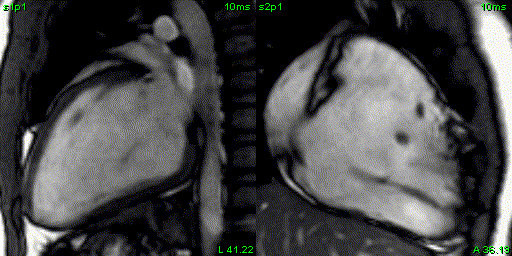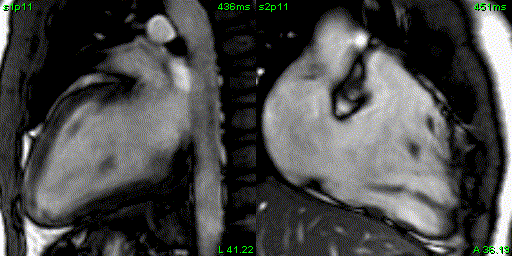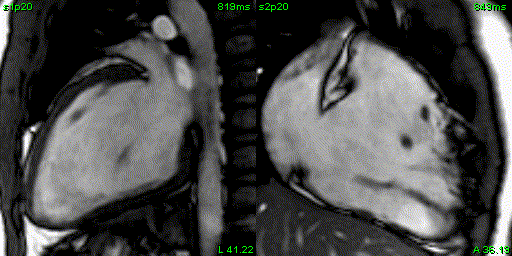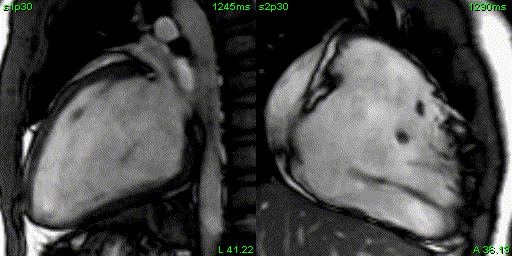
Figure 4: 2 chamber left ventricular and right ventricular SSFP cine.
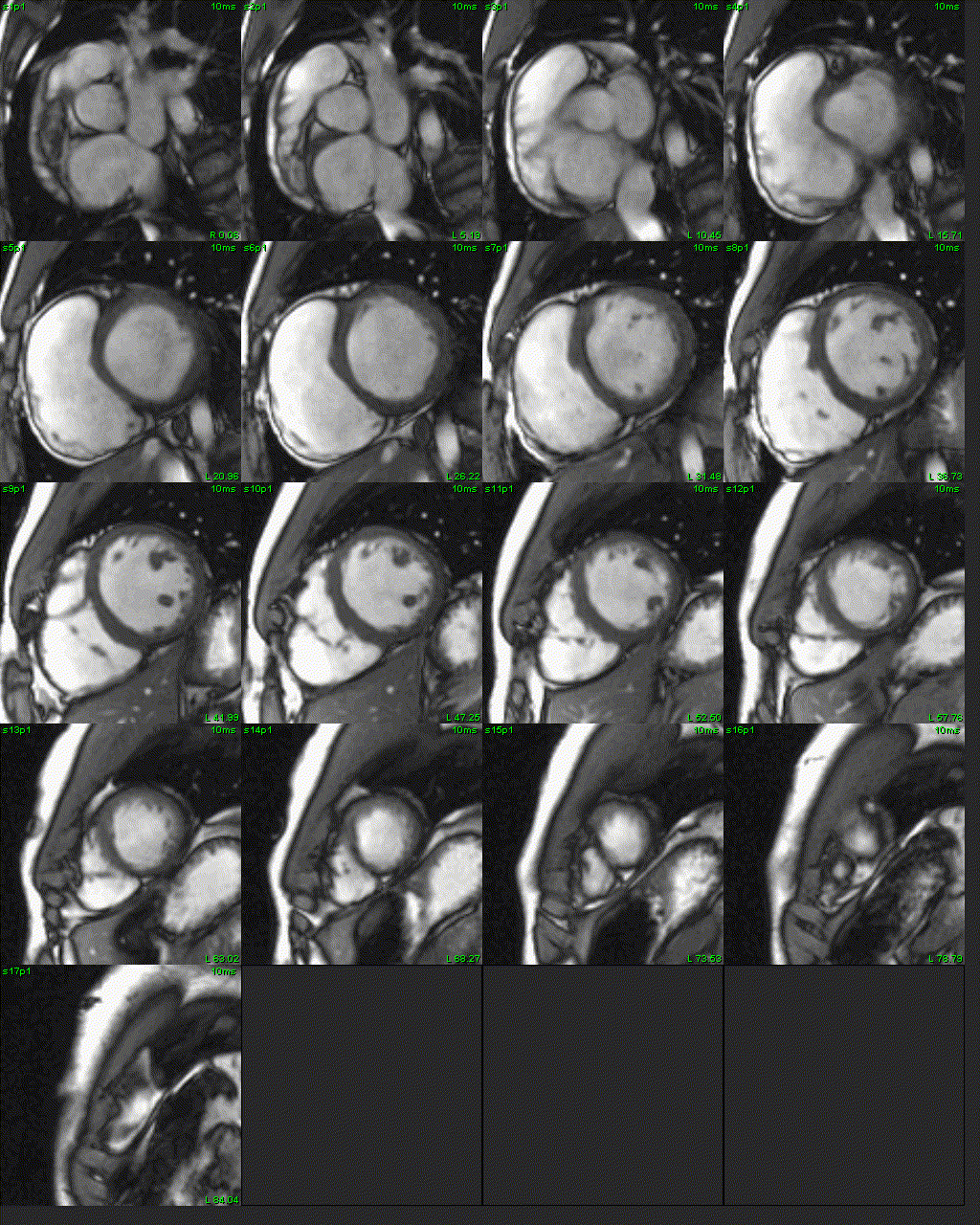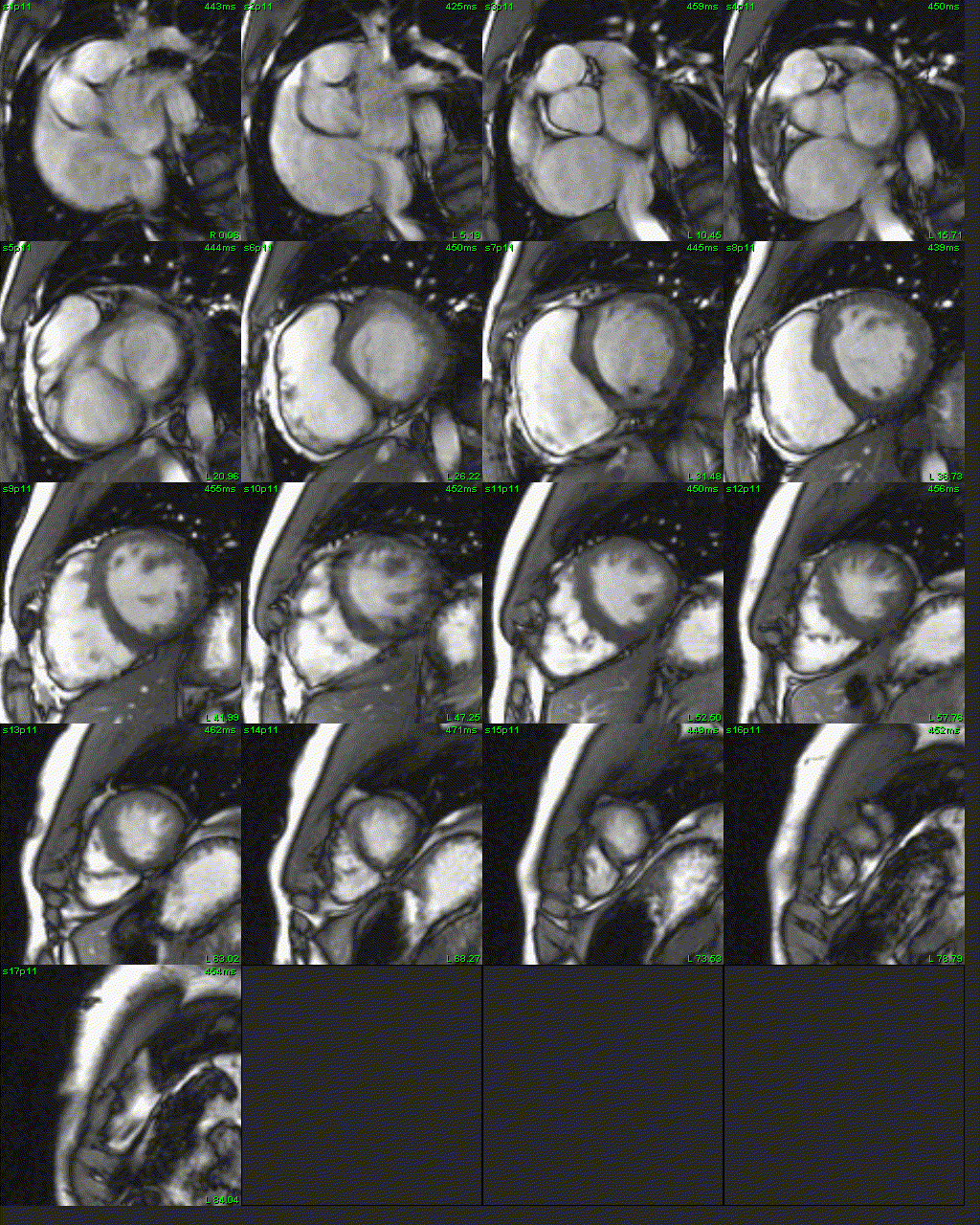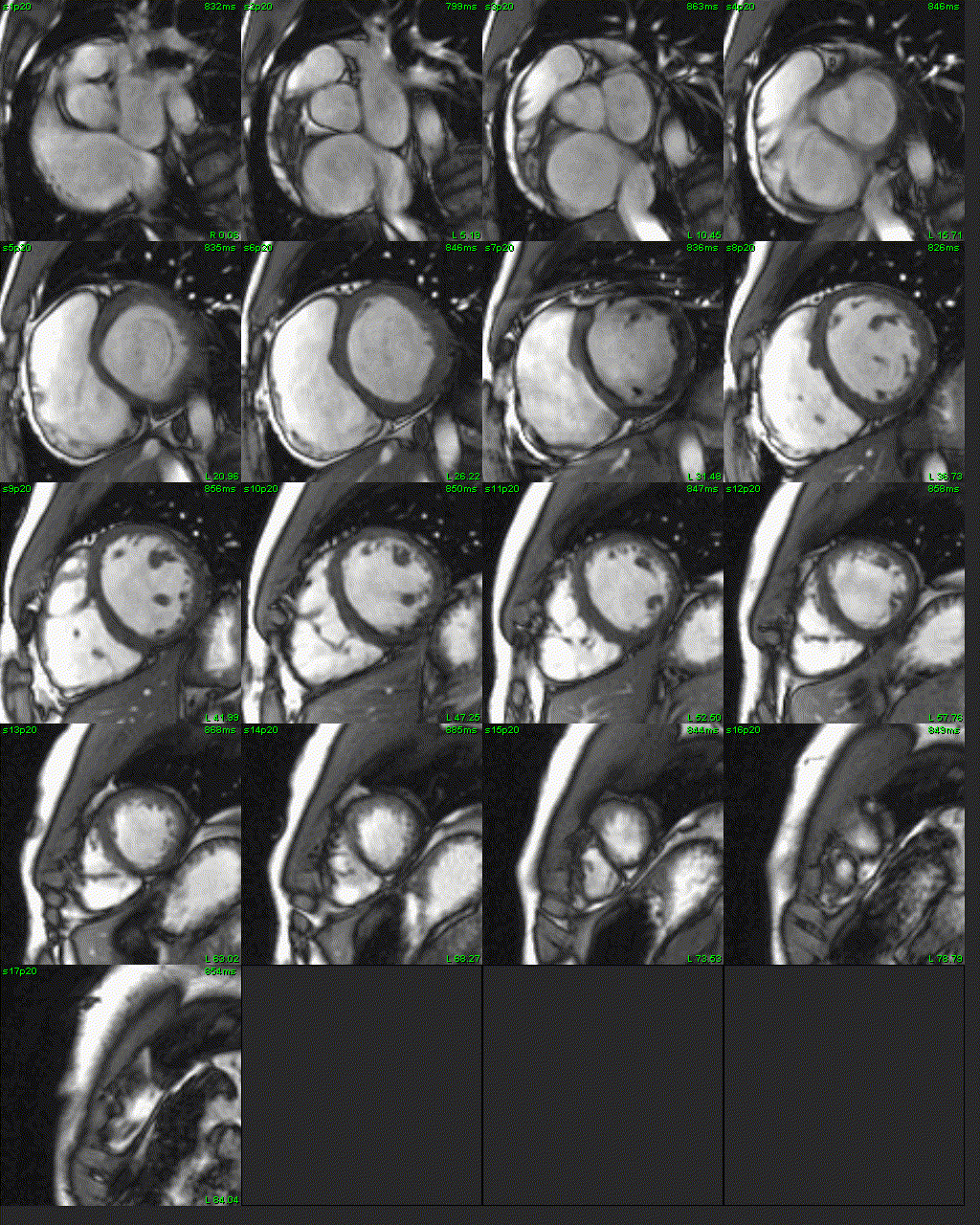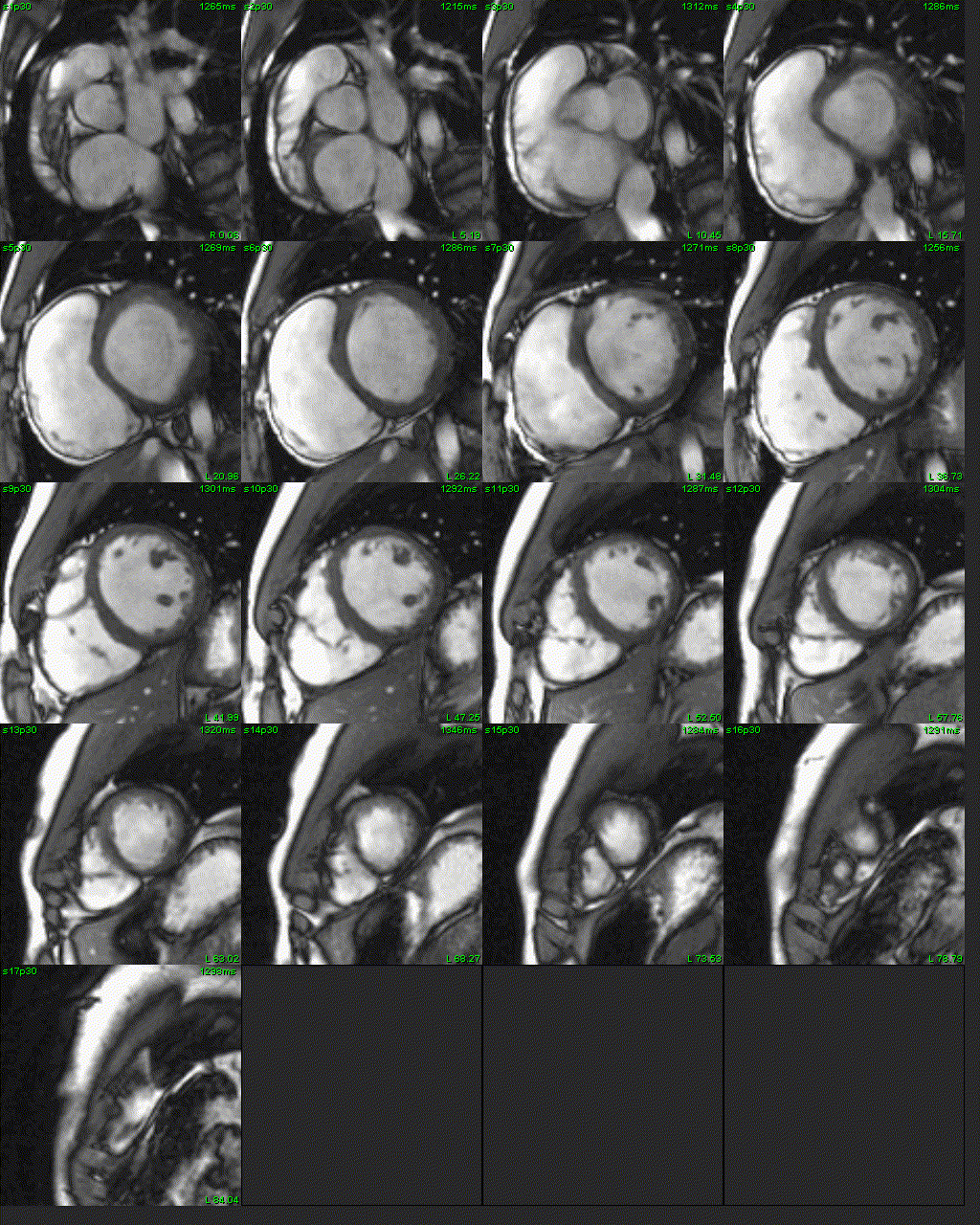
Figure 5: Short axis SSFP cine.
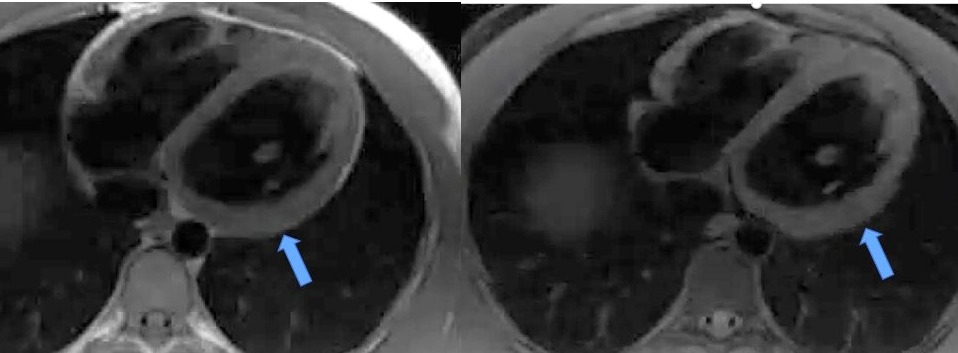
Figure 6: Black blood T1-weighted imaging with and without fat saturation.
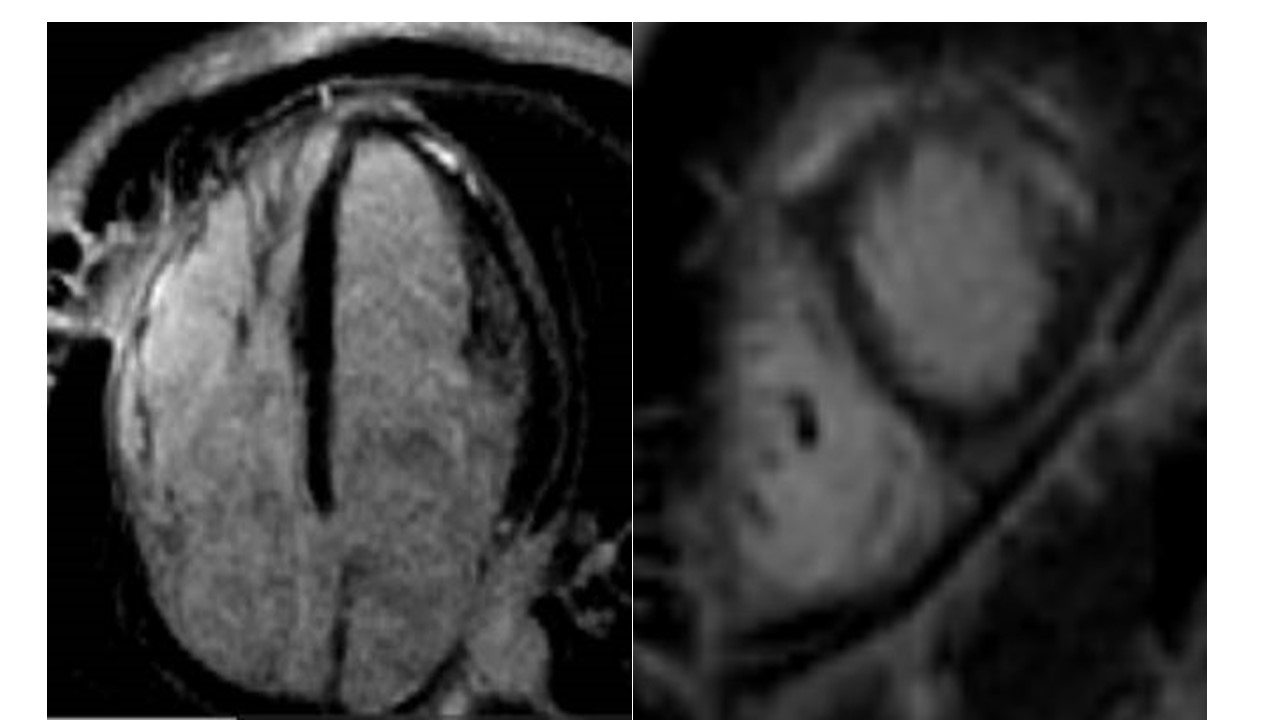
Figure 7: Late gadolinium enhancement imaging on 4-chamber (left) and short axis (right) views.
Conclusion
CMR findings fulfilled the major criteria for arrhythmogenic right ventricular cardiomyopathy (ARVC) developed by the 2010 ARVC Task Force 1. Recent case studies have demonstrated biventricular disease involvement is more common than previously reported, prompting the term ARVC to be relabeled as arrhythmogenic cardiomyopathy 2. His initial EKG showed monomorphic ventricular tachycardia suspicious for arrhythmogenic cardiomyopathy. To determine whether he required an implantable cardioverter defibrillator (ICD) alone vs. ICD placement and ventricular tachycardia ablation, he underwent an isoproterenol challenge test, which was positive. Subsequently, genetic testing found him to be positive for the desmoglein-2 (SG2) missense mutation. CMR demonstrated focal right ventricular dyskinesia with “accordion sign” and the right ventricular end-diastolic volume index measured greater than >110 mL/m2, with a right ventricular ejection fraction >40- < 45%. He thereby satisfied two major diagnostic criteria for arrhythmogenic cardiomyopathy with positive genetic testing and right ventricular findings on CMR. In addition, the presence of subepicardial to mid-wall late gadolinium enhancement at the left ventricular apex and possible left ventricular fibrofatty infiltration on T1-weighted imaging was consistent with arrhythmogenic cardiomyopathy with left ventricular involvement. He ultimately had an ICD placed and nadolol was initiated.
Perspective
Arrhythmogenic cardiomyopathy is a rare inherited disorder that causes fibrofatty replacement of cardiac tissue3-4. This is a known cause of cardiac death in young athletes, typically found in men less than 35 years old3. Presenting symptoms are generally non-specific and may include palpitations, fatigue, syncope, or even aborted sudden cardiac death. CMR is a valuable diagnostic tool that can identify arrhythmogenic cardiomyopathy with and without left ventricular involvement and guide clinical management. CMR diagnostic criteria were first established in 1994 and further revised in 20101. Initial subtle structural changes to the right ventricle can progress to involve both ventricles1. Tissue characterization is a vital tool in diagnosing arrhythmogenic cardiomyopathy, particularly in assessment of left ventricular involvement, which may not be apparent on echocardiography. Studies have shown that subepicardial left ventricular fatty infiltration, similar to our case, is a common finding in arrhythmogenic cardiomyopathy.5-6 With the autosomal dominant inheritance pattern, family history is a critical component to this diagnosis. Several genetic variations in the desmosomes have been found, such as the DSG2 seen in our patient. Some genetic variants are associated with an increased risk of malignant arrhythmias. While arrhythmogenic cardiomyopathy guidelines are evolving, the 2019 Heart Rhythm Society consensus statement has incorporated genetic data into the risk stratification for ICD implantation.7
This case exemplifies the importance of obtaining CMR for suspected arrhythmogenic cardiomyopathy, even in the presence of normal echocardiography. CMR imaging allowed for more detailed visualization of the ventricles and tissue characterization with parametric mapping and late gadolinium enhancement. CMR findings may prompt genetic workup, which in turn could inform prognosis and management for current and future family members.
Click here to view the entire study via CloudCMR.
Acknowledgments: We would like to thank Dr. Anjali Chelliah for her assistance with this case.
References
3. Anderson E. Arrhythmogenic right ventricular dysplasia. Am Fam Physician. 2006.73(8):1391-1398.
4. Basso C, Corrado D, Marcus F, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009.373(9671):11-17.
5. Riele A, Tandri H, Bluemke D. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J. Cardiovasc Magn Reason. 2014.16(50).
6. Corrado D, van Tintelen P, McKenna W, et al. Arrhythmogenic right ventricular cardiomyopathy evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020.41(14): 1414-1429
Case prepared by:
Dr Lee Jonan Chun Yin
Associate Editor, SCMR Case of the Week

