



Heart Center, Central Clinic Bad Berka, Germany
Clinical History
A 68-year-old woman presented with persistent and gradually progressing dyspnea (New York Heart Association Class III) and chronic cough, leading to suspicion of pulmonary sarcoidosis after a bronchoscopic examination. Subsequent echocardiography at the outpatient cardiologist revealed left ventricular hypertrophy (17 mm septal thickness) with preserved ejection fraction, prompting concern for cardiac sarcoidosis. A cardiovascular magnetic resonance (CMR) was recommended.
During her clinic visit, the patient reported only dyspnea and chronic cough, with no other relevant medical conditions except arterial hypertension. Despite suspicion of chronic obstructive pulmonary disease (COPD) /Asthma, pulmonary therapy had no effect. Notably, the patient had been aware of left ventricular hypertrophy for nearly a decade.
The clinical examination revealed no abnormalities. The electrocardiogram (ECG) showed a sinus rhythm without signs of left ventricular hypertrophy, but with T-wave inversion in inferolateral leads (Figure 1). Laboratory tests indicated mild iron deficiency anemia, elevated troponin levels (62 ng/l, normal range < 14 ng/l), and elevated NT-proBNP (609 pg/ml, normal range < 125 pg/ml).
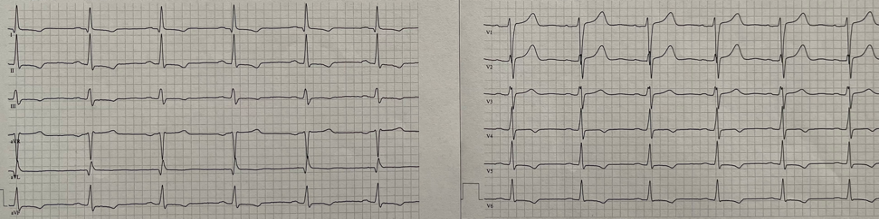 |
Figure 1. 12-lead ECG demonstrating sinus rhythm with T-wave inversion in the infero-lateral leads.
The initial CMR disclosed septal-predominant left ventricular hypertrophy with preserved systolic function without any regional motion abnormalities. Notably, globally normal and locally elevated T1 and T2 relaxation times were observed inferolateral at the basal level, along with intramural late gadolinium enhancement (LGE) in that region. These findings were interpreted as evidence of active focal myocardial inflammation, indicative of cardiac sarcoidosis.
The CMR findings were prompted by a positron emission tomography–computed tomography (PET-CT) scan, which revealed irregular, patchy to diffuse tracer accumulation in the left myocardium, particularly in the posterior wall, with a maximum standardized uptake value (SUVmax) of 16.4. This was interpreted as evidence of cardiac sarcoidosis (Figure 2).
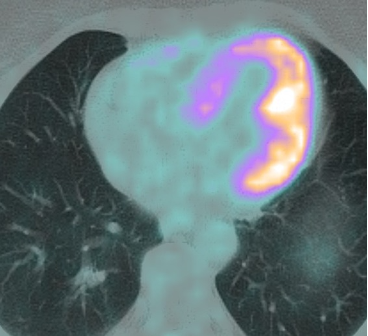 |
Figure 2. PET-CT demonstrating patchy tracer accumulation in the posterior/lateral LV wall.
A left ventricular biopsy was conducted, which provided inconclusive results, revealing minimal variation in cardiomyocyte nuclear size and isolated lymphocytes in the interstitium. Unfortunately, the procedure led to a stroke with left-sided hemiparesis, successfully treated with thrombolytic therapy. Subsequent comprehensive pneumological diagnostics ruled out pulmonary sarcoidosis.
Due to the low probability of cardiac sarcoidosis, specific therapy was not initiated. A recommended follow-up CMR conducted three months later revealed identical findings to the initial CMR from three months prior.
CMR Findings
CMR was performed on a 1.5 Tesla Siemens Magnetom Avanto Fit scanner. The left ventricle exhibits a normal size (LVEDVi 79 ml/m²; normal range 45-93 ml/m²), characterized by predominant septal hypertrophy (105 g/m²; normal range 30-59 g/m²) with a septum thickness of 16 mm and posterior wall thickness of 8 mm, while maintaining preserved systolic left ventricular function (LVEF 57%) without regional motion abnormalities. The right ventricle maintains a normal size (RVEDVi 80 ml/m²; normal range 48-104 ml/m²) and preserved systolic function (RVEF 60%).
In the native T1 maps, globally normal T1 times were observed (950 ms ± 62 ms), with pathologically elevated values in the inferolateral basal region up to 1051 ms. In the T2 maps, global T2 times were within the normal range (51 ms ± 5 ms), but pathologically increased in the inferolateral basal area up to 58 ms (Figure 3).
 |  |
Figure 3. Short-axis colormap of T1 (left) and T2 – relaxation times (right) at the basal level.
In the late contrast enhancement imaging, intramural late gadolinium enhancement (LGE) was evident in the inferolateral basal region (Figure 4). There were no indications for a left-ventricular thrombus.
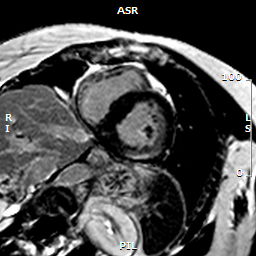 |
Figure 4. Short-axis LGE stack depicting epicardial enhancement in the basal inferolateral wall.
Conclusion
The second CMR raised suspicions of genetic hypertrophic cardiomyopathy. Subsequently, we sent an envelope containing a Fabry genetic test to the primary care physician. The results confirmed a positive diagnosis, identifying a heterozygous pathogenic variant in c.644A>G (p.Asn215Ser) and elevated lyso-Gb3 enzyme levels. As a result, chaperone therapy began following comprehensive diagnostics and clinical assessment.
Perspective
To the best of our knowledge, this case report represents the first published instance of a woman with an unidentified heterozygous alpha-galactosidase gene mutation, where the initial misinterpretation of left ventricular hypertrophy and myocardial inflammation in CMR and PET-CT as cardiac sarcoidosis was rectified through repeated CMR and a comprehensive review of results in the clinical context, ultimately leading to the diagnosis of Fabry disease.
Fabry disease, a rare genetic disorder due to X-chromosomal inheritance, results from alpha-galactosidase A enzyme deficiency, causing the accumulation of Globotriaosylceramide (Gb3) in tissues. Recent studies challenge the notion that it predominantly affects males, revealing diverse clinical symptoms in heterozygous Fabry women as well with a single mutated GLA gene as well. Impacting both genders, it presents classic and late-onset subtypes. The classic phenotype, marked by minimal alpha-galactosidase A activity, shows early symptoms like pain, skin changes, kidney problems, and cardiovascular complications. Conversely, the late-onset phenotype, usually linked with residual Gal A activity, presents delayed or single-organ manifestations, such as cardiac issues, complicating diagnosis[1].
When dealing with a female patient suspected of having Fabry disease, two considerations must be made based on clinical evidence. Fabry disease in heterozygous females tends to manifest clinically more frequently as a late onset Fabry, with a highly heterogeneous and mild course, but often with a dominant cardiac involvement.[2] The primary manifestation of cardiac involvement is left ventricular hypertrophy, often associated with heart failure symptoms. This can be observed in up to 2/3 of heterozygous female patients in advanced age.[3] Additionally, it is crucial to emphasize that in adult patients with unexplained left ventricular hypertrophy, Fabry disease can be identified with an incidence of up to 1%.[4]
In classic teachings, the passive accumulation of sphingolipids in myocardial tissues triggers left ventricular hypertrophy, valve defects, chronic ischemia, and increased fibrosis. Recent studies propose that glycolipids are not only passive stored in organs but also trigger a chronic pro-inflammatory response. In other words, it is not just a storage disease causing hypertrophic cardiomyopathy but rather an inflammatory hypertrophic cardiomyopathy.[5]
CMR findings in Fabry cardiomyopathy significantly vary with the disease stage. In the early stages, there is predominant progressive concentric left ventricular hypertrophy with globally reduced T1 relaxation times, attributed to lipid accumulation in the myocardium. Typical findings in later stages of disease progression include replacement fibrosis on the basal posterior wall, visible on LGE as a non-ischemic scar. There may be a tendency towards asymmetric septal hypertrophy due to thinning of the posterior wall from chronic inflammatory scarring. This is often accompanied by pseudo-normalization of T1 and T2 maps due to progressive interstitial fibrosis and inflammation, and even pathological elevations of the T1 and T2 relaxation times in scarred regions. The intricacy of these manifestations presents diagnostic challenges, as demonstrated in our case of advanced late-onset Fabry cardiomyopathy.[6]
There is increasing evidence supporting the utility of 18F-Fluorodeoxyglucose Positron Emission Tomography-computed Tomography (18F-FDG-PET) in patients with known Fabry cardiomyopathy. The PET findings correlate well with the disease stage and treatment response. Myocardial inflammation observed in PET aligns closely with the areas of late gadolinium enhancement (LGE) on LGE-MRI, suggesting that LGE in Fabry cardiomyopathy is not solely a result of scarring but also involves inflammation, as demonstrated in our case. It’s worth noting that PET findings of myocardial inflammation, as observed in our case, can be non-specific and should not independently guide clinical diagnosis.[7]
The genetic test in our patient confirmed the diagnosis of Fabry Disease. Current data suggests that up to one-third of patients diagnosed with cardiac sarcoidosis through imaging studies show pathological mutations in genetic testing indicative of genetic cardiomyopathy, resembling inflammatory cardiomyopathy. These findings advocate for the integration of genetic testing into the diagnostic workup of patients with cardiomyopathy accompanied by myocardial inflammation, enabling a more precise characterization of such patients.[8]
In summary, highlighting the importance of interpreting the CMR findings within a clinical context was crucial for accurately diagnosing Fabry cardiomyopathy in this intricate case. Clinical diagnosis of specific cardiac conditions like sarcoidosis based solely on isolated imaging findings should be avoided.
References
- Pieroni M, Moon JC, Arbustini E, Barriales-Villa R, Camporeale A, Vujkovac AC, Elliott PM, Hagege A, Kuusisto J, Linhart A, Nordbeck P, Olivotto I, Pietilä-Effati P, Namdar M. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J Am Coll Cardiol. 2021 Feb 23;77(7):922-936. doi: 10.1016/j.jacc.2020.12.024.
- Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, Brühl K, Gal A, Bunge S, Beck M. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001 Dec;24(7):715-24. doi: 10.1023/a:1012993305223.
- Kampmann C, Baehner F, Whybra C, Martin C, Wiethoff CM, Ries M, Gal A, Beck M. Cardiac manifestations of Anderson-Fabry disease in heterozygous females. J Am Coll Cardiol. 2002 Nov 6;40(9):1668-74. doi: 10.1016/s0735-1097(02)02380-x.
- Elliott P, Baker R, Pasquale F, Quarta G, Ebrahim H, Mehta AB, Hughes DA; ACES study group. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson-Fabry Disease survey. Heart. 2011 Dec;97(23):1957-60. doi: 10.1136/heartjnl-2011-300364. Epub 2011 Sep 2.
- Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017 Nov;122(3):19-27. doi: 10.1016/j.ymgme.2017.09.004. Epub 2017 Sep 13.
- Nordin S, Kozor R, Medina-Menacho K, Abdel-Gadir A, Baig S, Sado DM, Lobascio I, Murphy E, Lachmann RH, Mehta A, Edwards NC, Ramaswami U, Steeds RP, Hughes D, Moon JC. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC Cardiovasc Imaging. 2019 Aug;12(8 Pt 2):1673-1683. doi: 10.1016/j.jcmg.2018.03.020. Epub 2018 May 16.
- Nappi C, Ponsiglione A, Pisani A, Riccio E, Di Risi T, Pieroni M, Klain M, Assante R, Acampa W, Nicolai E, Spinelli L, Cuocolo A, Imbriaco M. Role of serial cardiac 18F-FDG PET-MRI in Anderson-Fabry disease: a pilot study. Insights Imaging. 2021 Sep 6;12(1):124. doi: 10.1186/s13244-021-01067-6.
- Subramanian M, Ravikanth VV, Saggu D, Yalagudri S, Narasimhan C. Genetic Cardiomyopathies Misdiagnosed as Cardiac Sarcoidosis. JACC Clin Electrophysiol. 2024 Mar;10(3):583-584. doi: 10.1016/j.jacep.2023.12.010.
Case prepared by:
Pranav Bhagirath, MD, PhD
Editorial Team, Cases of SCMR
Department of Cardiology
Amsterdam University Medical Centers
Amsterdam, The Netherlands