



Department of Medicine, Cardiology, Brooke Army Medical Center, Fort Sam Houston, TX
Clinical History
A 49-year-old previously healthy active-duty female with past medical history of hypohydrosis, iron deficiency anemia with hyperferritinemia and three miscarriages presented for evaluation of exertional dyspnea and an inability to complete a military fitness test. She also endorsed palpitations, chest pressure and peripheral neuropathy. Vitals were within normal limits, and the physical exam was unremarkable. Laboratory evaluation was within normal limits and showed no evidence of iron overload (Table 1). Electrocardiogram (ECG) was significant for left ventricular hypertrophy (LVH) with strain pattern and left atrial enlargement (Figure 1).
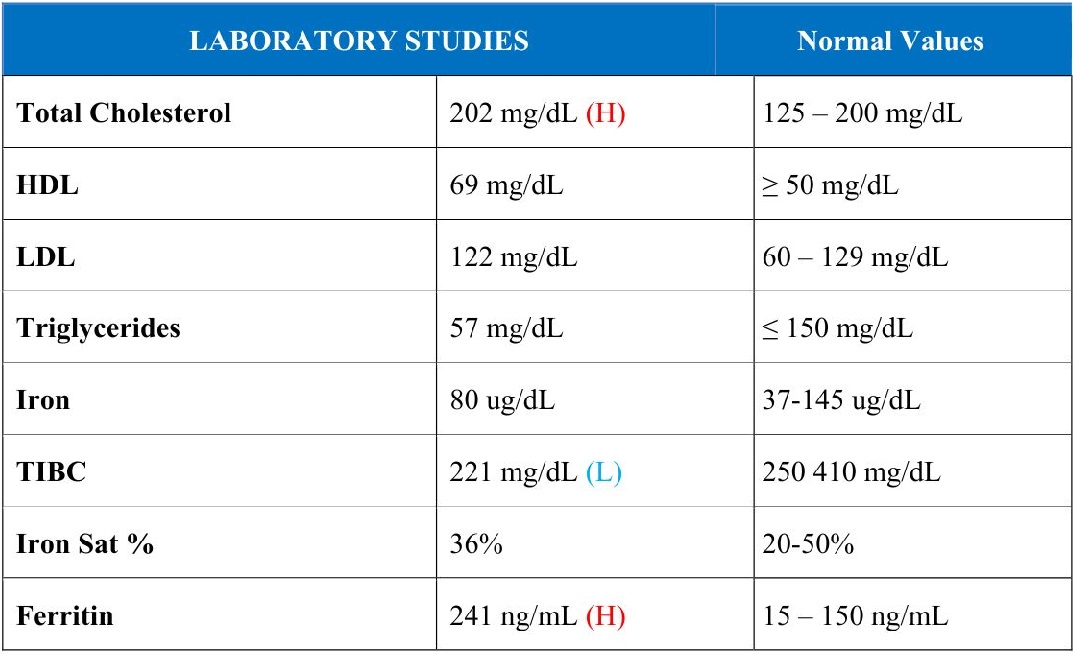 |
| Table 1: Initial laboratory values demonstrating no evidence of iron overload. HDL = high density lipoprotein; LDL = low density lipoprotein; TIBC = total iron binding capacity. |
 |
| Figure 1: Electrocardiogram (ECG) with left ventricular hypertrophy (LVH) with strain and left atrial enlargement (LAE). |
The differential diagnosis included hypertension, athlete’s heart, coronary artery disease, aortic stenosis, amyloidosis, hypertrophic cardiomyopathy (HCM), and Fabry Disease.
A transthoracic echocardiogram (TTE) showed increased wall thickness with apical hypertrophy and diastolic dysfunction. There was no significant right ventricular systolic or valvular dysfunction (Figure 2).
 |
| Figure 2: Transthoracic echocardiogram: Apical hypertrophy. (A) Parasternal Long axis. (B) Apical Four Chamber. (C) Apical Two Chamber. (D-E) Apical Four Chamber in diastole, then systole with echo enhancing agent. |
She completed an exercise treadmill test, achieving 10 minutes on a Bruce protocol (12.1 Metabolic Equivalents of Tasks) correlating with good functional capacity and had no evidence of exercise-induced arrhythmias or symptoms. A coronary computed tomography angiography (CorCTA) revealed no evidence of coronary atherosclerosis or obstructive coronary disease and no evidence of any other associated aortopathy in the visualized portions of the thoracic aorta (Figure 3).
 |
| Figure 3: CorCTA with no coronary artery disease. A: Left anterior descending artery, B: Left circumflex artery, C: Right coronary artery. |
CMR Findings
Initial Cardiac Magnetic Resonance Imaging (CMR) was obtained without parametric mapping, and she was diagnosed with apical variant non-obstructive HCM. She proceeded with guideline recommended risk stratification for HCM. However, upon re-evaluation nine months later, she was found to have a strong paternal family history of Fabry Disease. Genetic testing was obtained and significant for a pathologic GLA gene variant c.861G>T p.(Trp287Cys), consistent with Fabry disease. A repeat CMR using a 1.5 Tesla Avanto Fit (Siemens Healthineers AG, Erlangen, Germany) magnet was completed with parametric mapping. Findings included a normal left ventricular ejection fraction of 69.3% (normal: 52-79%), normal end-diastolic volume of 71 mL/m2 (normal: 50-96 mL/m2), and asymmetric wall thickness predominantly in the apical segments (anterior, septal, inferior, and lateral) with up to 16 mm in the apical lateral wall. There was also patchy mid-wall late gadolinium enhancement (LGE) of the apical segments, abnormally low native T1 map times involving the basal-mid segments (base: 855 ms and apex: 938 ms), and extracellular volume (ECV) of 28% [Figures 4-6, Movie 1].
 |
| Figure 4: CMR cine balanced steady state free precession (bSSFP) at diastole in basal (A), mid (B), apical (C) short axis, two chamber (D), four chamber (E), and three chamber (F) with apical hypertrophy present. |
 |
| Movie 1: Cine 4 chamber bSSFP demonstrating apical left ventricular hypertrophy and normal biventricular systolic function. |
 |
| Figure 5: CMR with late gadolinium enhancement (LGE) Orange Arrows. (A-B) Apical 2 Chamber. (C) Apical 4 Chamber. (D) Short Axis: Apical. |
 |
| Figure 6: CMR with T1 parametric mapping revealing low T1 map times consistent with Fabry Disease (Color Scale). (A) Short Axis: Base. (B) Short Axis: Mid. (C) Short Axis: Apical. (D) Four Chamber. (E) Two Chamber. |
Conclusion
The patient officially was diagnosed with Fabry Disease and initiated on enzyme replacement therapy with biweekly agalsidase beta. She improved clinically. She does not have exercise restrictions and is now able to play pickleball, golf 9 holes and cycle. She is currently undergoing a medical evaluation board evaluation for discharge from active-duty service. She will require enzyme replacement lifelong or at least until gene replacement therapy is available. Current clinical trials for gene replacement therapy are in progress.
The accurate and timely diagnosis of Fabry Disease in female heterozygotes is of critical importance. Nevertheless, misdiagnosis remains commonplace in female patients presenting with LVH. Parametric mapping on CMR is key in differentiating between Fabry Disease and other cardiomyopathies with LVH as a characteristic feature, and it should be included in the CMR protocol for the evaluation of this disease.
Perspective
Fabry disease is a rare, X-linked lysosomal storage disease caused by a mutation in the GLA gene resulting in the absence or deficiency in the α-galactosidase enzyme. Absence of this enzyme results in progressive neural glycosphingolipid accumulation within lysosomes of vascular endothelial and smooth-muscle cells throughout the body.[3] Fabry disease in female heterozygotes historically has been thought of clinically silent or a merely mild disease compared to male hemizygotes. However, Fabry disease has been shown to present with more serious clinical manifestations in females than previously thought.[1] Compared to males, female patients with Fabry disease and cardiac involvement tend to present later in life with findings of left ventricular hypertrophy, conduction abnormalities, arrhythmias, ascending aorta dilatation, coronary artery disease, and valvular disease.[2] In a minority of patients, the clinical manifestations of Fabry disease are confined to the myocardium.[3] A high index of clinical suspicion and the appropriate use of multimodality cardiac imaging are critical to the accurate and timely diagnosis of Fabry disease.
In classic cases, affected male patients have no detectable α-galactosidase activity, resulting in severe clinical manifestations in childhood or adolescence. In male patients with atypical variants and in female heterozygotes who have some residual α-galactosidase activity, clinical manifestations tend to be attenuated, late-onset, and involve the myocardium, cerebrum, and renal vasculature. Notably, α-galactosidase enzyme activity in females with Fabry disease with severe cardiac manifestations can be normal, necessitating genetic testing for accurate diagnosis.[2]
The most common cardiac abnormality in female patients with Fabry disease is LVH affecting approximately 25% of patients at a mean age of 52 years.[4] LVH increases in both prevalence and severity with age, affecting nearly all female patients with Fabry disease above age 45.[2] In patients with late onset LVH, Fabry disease may account for up to 12% of cases.[5] Though concentric hypertrophy is the most common pattern seen in Fabry disease, it can also mimic the structural characteristics of various subtypes of hypertrophic cardiomyopathy, including asymmetric septal and apical variants, as seen in this case.[6]
The treatment of Fabry disease involves enzyme replacement therapy with either agalsidase alfa or beta to restore absent or deficient levels of the α-galactosidase enzyme. Enzyme replacement therapy has shown to stabilize and even decrease left ventricular mass and wall thickness, as well as improve quality of life.[10]
CMR is a crucial imaging modality in identifying Fabry disease and differentiating it from HCM and other cardiomyopathies with LVH as a characteristic feature. Typical findings of Fabry disease on CMR include concentric LVH and basal to mid inferolateral wall LGE.[7] In our case, the patient was found to have patchy mid-wall LGE of the apical segments, highlighting how patterns found in females can vary more so than in classic male examples. Both LVH and LGE are strong predictors of adverse cardiac events in Fabry diease.[8] Parametric mapping on CMR includes T1 mapping (native and post-contrast), T2 mapping, and extracellular volume assessment. In Fabry disease specifically, studies on parametric mapping have shown a reduction in native T1 times and high regional T2 relaxation times in areas of LGE, consistent with inflammation.[9] Comparatively, HCM demonstrates delayed enhancement within the regions of hypertrophy and shows increased native T1 times.[9]
Disclaimer: The views expressed are those of the author(s) and do not necessarily reflect the official policy or position of the Defense Health Agency, Brooke Army Medical Center, the Department of Defense, nor any agencies under the U.S. Government.
Acknowledgements: Stephanie A. Howes, DO, Serafim Perdikis, MD
Click here to see the entire case on CloudCMR.
References
- Izhar, Raafiah et al. “Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations.” Genes vol. 15,1 37. 26 Dec. 2023, doi:10.3390/genes15010037.
- Kampmann, Christoph et al. “Cardiac manifestations of Anderson-Fabry disease in heterozygous females.” Journal of the American College of Cardiology vol. 40,9 (2002): 1668-74. doi:10.1016/s0735-1097(02)02380-x.
- von Scheidt, W et al. “An atypical variant of Fabry’s disease with manifestations confined to the myocardium.” The New England journal of medicine vol. 324,6 (1991): 395-9. doi:10.1056/NEJM199102073240607.
- Deegan, Patrick B, et al. “Fabry disease in females: clinical characteristics and effects of enzyme replacement therapy.” Fabry Disease: Perspectives from 5 Years of FOS, edited by Atul Mehta et. al., Oxford PharmaGenesis, 2006.
- Chimenti, Cristina et al. “Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy.” Circulation vol. 110,9 (2004): 1047-53. doi:10.1161/01.CIR.0000139847.74101.03.
- Deva, Djeven Parameshvara et al. “Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in Anderson-Fabry disease.” Journal of cardiovascular magnetic resonance : official journal of the Society for Cardiovascular Magnetic Resonance vol. 18 14. 31 Mar. 2016, doi:10.1186/s12968-016-0233-6.
- De Cobelli, Francesco et al. “Delayed-enhanced cardiac MRI for differentiation of Fabry’s disease from symmetric hypertrophic cardiomyopathy.” AJR. American journal of roentgenology vol. 192,3 (2009): W97-102. doi:10.2214/AJR.08.1201.
- Hanneman, Kate et al. “Left Ventricular Hypertrophy and Late Gadolinium Enhancement at Cardiac MRI Are Associated with Adverse Cardiac Events in Fabry Disease.” Radiology vol. 294,1 (2020): 42-49. doi:10.1148/radiol.2019191385.
- Kottam, Anupama et al. “State-of-the-Art Imaging of Infiltrative Cardiomyopathies: A Scientific Statement From the American Heart Association.” Circulation. Cardiovascular imaging vol. 16,11 (2023): e000081. doi:10.1161/HCI.0000000000000081.
- Azevedo, Olga et al. “Fabry Disease Therapy: State-of-the-Art and Current Challenges.” International journal of molecular sciences vol. 22,1 206. 28 Dec. 2020, doi:10.3390/ijms22010206.
Case prepared by:
Robert D. Tunks, MD, MHS
Editorial Board Member, Cases of SCMR
Penn State Health Children’s Hospital, Hershey Pennsylvania USA