



Case from: Jonathan Windram, MBChB1,2, Timothy J. Bradley, MBChB1, Shi-Joon-Yoo, MD1,2, Lars Grossse-Wortmann, MD1,2
Institute: 1Labatt Family Heart Centre at The Hospital for Sick Children, University of Toronto; Toronto, Ontario, Canada, 2 Department of Diagnostic Imaging, The Hospital for Sick Children, University of Toronto; Toronto, Ontario, Canada.
Clinical History:
A four week old male infant was noted to be dysmorphic including hypertelorism, bilateral inguinal hernias, an umbilical hernia and bilateral clubfeet. A right axillary mass was noted on examination; ultrasound revealed the mass to be a dilated and tortuous vessel but could not conclude definitively whether it was venous or arterial.
An echocardiogram revealed a dilated aortic root and main pulmonary artery and a large ductal aneurysm (Movie 1). The pulmonary valve was thickened and quadricuspid. A cardiac magnetic resonance (CMR) scan was performed to further delineate the anatomy.
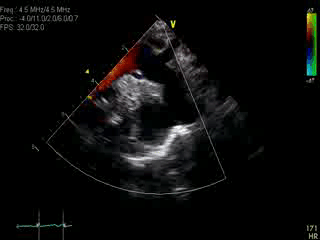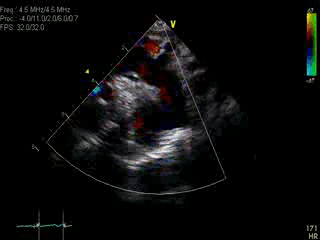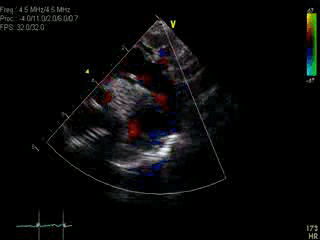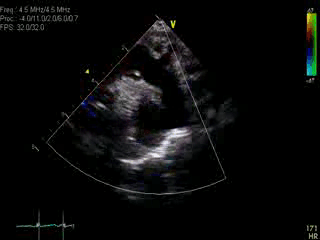
Movie 1: Transthoracic echocardiographic image with color Doppler showing the dilated aorta with a large ductal aneurysm.
CMR Findings:
The situs was solitus with levocardia, the intracardiac anatomy was normal with concordant atrio-ventricular and ventriculo-arterial connections.
The aorta was enlarged and tortuous (Figure 1). The distal thoracic aorta crossed the midline from right to left and then back again just above the diaphragm, forming an acute angle. The caliber of the aorta was heterogeneous throughout its course, creating an irregular appearance. The branching pattern was normally sequenced but all of the head and neck vessels were noted to be tortuous with a cork-screw appearance. The right innominate artery turned back upon itself. Both vertebral arteries were small and very tortuous, whereas the remainder of the head and neck vessels was dilated. The subclavian and axillary arteries were aneurysmally enlarged, especially on the right which accounted for the mass noted in the axilla.
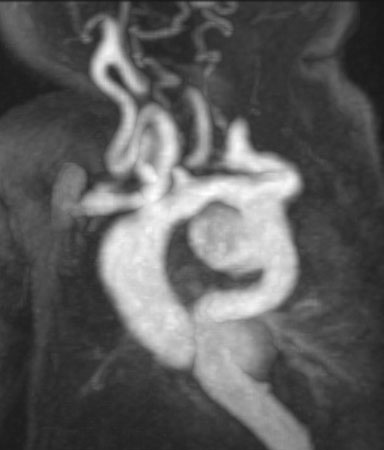
Figure 1: Coronal maximum intensity projection of a magnetic resonance angiogram showing an aneurysmal ascending aorta with tortuous head and neck vessels. The innominate artery “dips down” below the arch of the aorta.
There was a large ductal aneurysm, measuring 25 x 25 mm in cross section and 32 mm in length (Figure 2). Qualitatively, there was normal biventricular systolic function. The origins and proximal segments of the celiac trunk and superior mesenteric artery were mild to moderately ectatic (Movie 2). The renal, common iliac, and external iliac arteries were all tortuous and dilated.
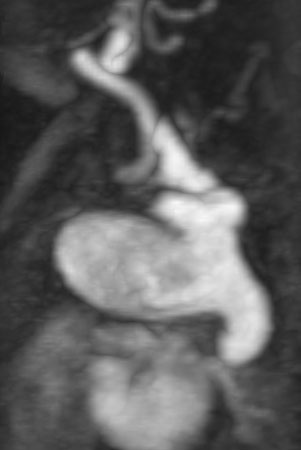
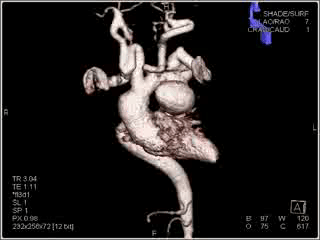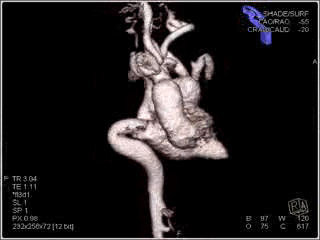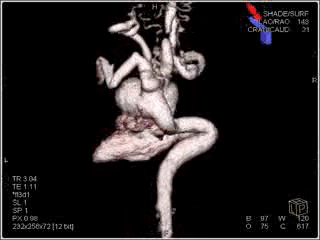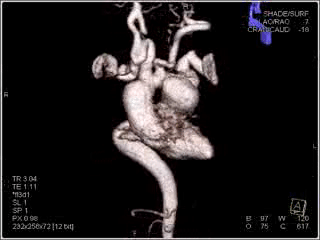
Figure 2: Sagittal view showing the ductal aneurysm as it arises from the descending aorta.
Movie 2: Volume rendered image of the aorta and branching arteries
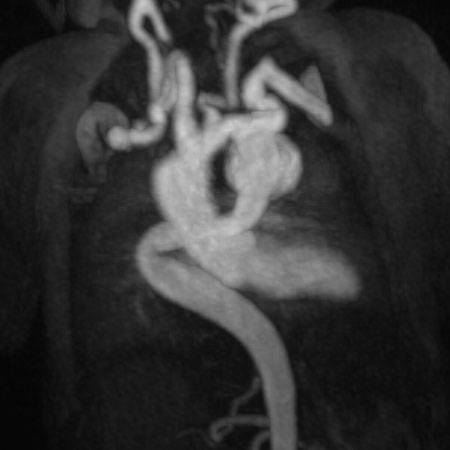
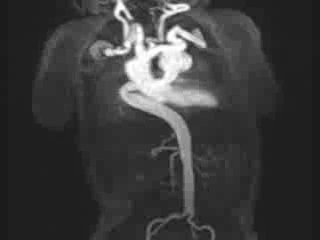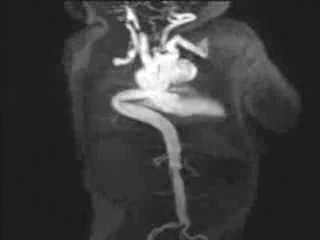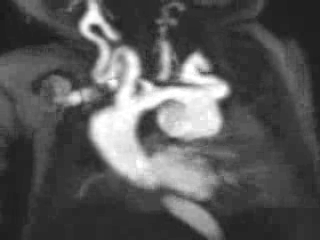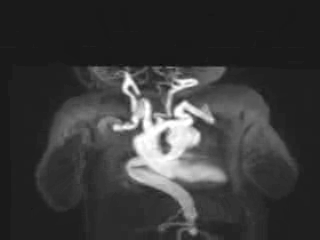
Figure 3: Maximum intensity projection image showing the aorta and branching arteries.
Movie 3: Maximum intensity image delineating the entire vascular anatomy.
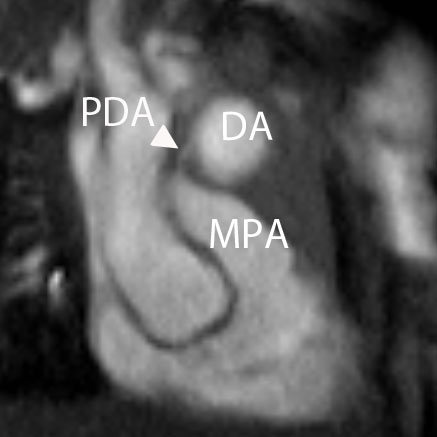
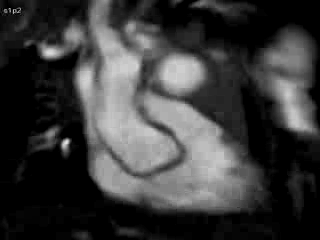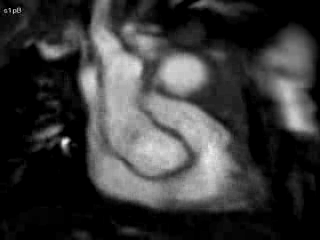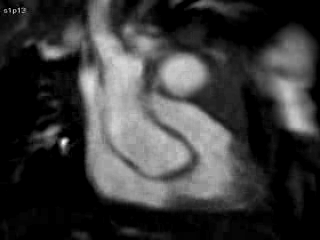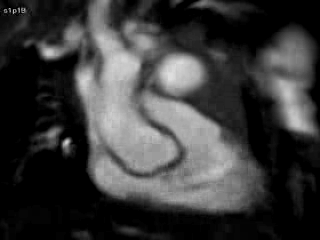
Figure 4 and Movie 4: Cine two chamber view of the heart showing a small persistent patent ductus arteriosus (PDA) communicating between the main pulmonary artery (MPA) and the ductal aneurysm.
Conclusion:
Based upon the CMR findings and the clinical features, a provisional diagnosis of Loeys-Dietz syndrome (LDS) was made which was subsequently confirmed by genetic studies.
Perspective:
LDS in a relatively recently recognized inherited connective tissue disorder. It results from autosomal dominantly inherited heterozygous mutations in the genes encoding for the transforming growth factor beta receptors 1 and 2, on chromosome 9q22 and 3p22, respectively (1). The exact prevalence is as yet unknown but may be greater than first suspected as there is a phenotypic overlap with other connective tissue disorders, likely causing some LDS patients to be misdiagnosed.
LDS overall carries a worse prognosis than Marfan syndrome with a median survival of 37 years (treated Marfan syndrome has a median survival of 70 years), although there is significant clinical heterogeneity (1). Aortic dissection or rupture of the aortic root is the most common cause of death in LDS. Aneurysmal dilation of the aorta has been described on fetal ultrasound (2) and there is one reported case of dissection occurring in a child aged 3 months (3). Early recognition of this condition is vital to ensure that close supervision is performed and surgery is undertaken, if necessary (1, 4, 5).
For an up to date review of the imaging findings of LDS, the reader is directed to the recent excellent review from Kalra et al. (6).
References:
- Loeys BL, Schwarze U, Holm T et al. (2006) Aneurysm syndromes caused by mutations of the TGF-beta receptor. N Engl J Med 355:788-798.
- Viassolo V, Lituania M, Marasini M et al. (2006) Fetal aortic root dilatation: a prenatal feature of the Loeys Dietz syndrome. Prenat Diagn 26:1081-1083.
- Malhotra A, Westesson PL. (2009) Loeys-Dietz syndrome. Pediatr Radiol 39:1015.
- Muramatsu Y, Kosho T, Magota M et al. (2010) Progressive aortic root and pulmonary artery aneurysms in neonate with Loeys-Dietz syndrome type 1B. Am J Med Genet A 152A:417-421.
- Yetman AT, Beroukhim RS, Ivy DD, et al. (2007) Importance of the clinical recognition of Loeys-Dietz syndrome in the neonatal period. Pediatrics. 119:e1199-202.
- Kalra V, Gilbert JW, Malhotra A. (2011) Loeys-Dietz syndrome: cardiovascular, neuroradiological and musculoskeletal imaging findings. Pediatr Radiol [Epub ahead of print].
COTW handling editor: M. Jay Campbell, MD
Have your say: What do you think? Latest posts on this topic from the forum

